
Everybody has woken up in the morning haunted by the question “how relevant is what I do going to be in the future?”. In case of chemists, all of us wonder what is the future of chemistry sometimes.
The answer to this question is especially relevant to the younger generations. Will I be able to find a job in 20 years doing exactly what I do now? Am I focusing on a branch of science that will be important in a couple of decades?
For now, it is impossible to predict the future of chemistry. However, it seems very likely that any field of science will evolve significantly with the advances on artificial intelligence.

And this is most likely not avoidable. Computers and robots are here to stay, and they are only getting better. But how much better can they become in our lifespan?
Artificial Intelligence and Machine Learning
Probably one of the biggest revolutions in science is the appearance of computers. Something that today we take for granted, has pushed the speed of scientific discovery for the past decades.
Today we almost cannot conceive synthetic chemistry without tools such as SciFinder or Reaxys. But how long until you can input a molecule that has never been made before in a search box, and you get exactly the steps you need to take to make it in a lab? Artificial intelligence (AI) and machine learning might be behind this through the future of chemistry.
If you are not familiar with those terms take two minutes to watch the video below:
You can teach a computer how to differentiate a cat from a dog.
Using AI to solve this problem is not very useful, since humans are already pretty good at it. However, when it comes to analyzing hundreds or thousands of data points at the same time, human beings are drastically outperformed by computers.
And, in a way, chemistry is a lot about this.
How Can the Future of Chemistry Be Defined by AI?
When you want to optimize a new synthetic step, or the properties of a new material, what do you usually do?
Dive into SciFinder, download a couple of reviews and 10 research papers, skim over the schemes, and from that, extrapolate the conditions you want to test in the lab.
Seeing it from an AI perspective, this procedure is extremely rudimentary, to say the least. And yet it’s what >90% of experimental chemists (as myself) do on a daily basis. And computers will eventually be better at it, no doubt about that.
Obviously there is a significant creativity component of a research jobs. And identifying or dealing with unknown results. At the early stage of AI and machine learning in which we are at, humans still outperform machines. It is difficult to know how long it will stay this way. Honestly, I’d be surprised if it took more than 10–15 years.
The Future of Chemistry Now
AI has been around for several decades. Getting better and better every day.
Computer-assisted chemical synthesis was pioneered by E. J. Corey as back as 1985, when he reported in Science a very basic system for synthetic analysis in organic chemistry. This was 5 years before being awarded his Nobel prize, in 1990, but there was not a big follow up on this kind of chemistry until more recently.
However, in the past couple of years, an explosion in computer-assisted chemistry is only getting started. This has been commented by F. Peiretti and J. M. Brunel in 2018, but even since that day, many more recent works have seen the light of day. This might indeed define the future of chemistry.
Some of the key players on this approaches are Abigail Doyle, Matt Sigman, Lee Cronin, or the MIT team led by Timothy Jamison and Klavs Jensen. We will try to briefly review some of the very last years.
We apologize in advance in case we missed some important work. This is not intended to be a comprehensive review, but rather just a collection of some examples to illustrate the idea.
Parameterization and Prediction
The research group led by Matthew Sigman at the University of Utah has plenty of collaborative projects based upon parameterization and prediction.
This method is based on applying predictive statistical analysis to chemical reactions. They part from MM and DFT calculations to abstract properties or parameters of ligands or catalysis. Then they run statistics comparing these parameters to the experimental results obtained with each ligand or catalyst. They come up with models that allow predicting how other ligands, catalysts or substrates would behave.
It apparently it works!
A recent example is a collaboration with Mark Biscoe, in which they show how ligand parameterization allows finding the best ligands to perform a enantiodivergent (you can choose the enantiomer you want as product just by tuning the ligand) Pd-catalyzed C–C cross coupling reaction.
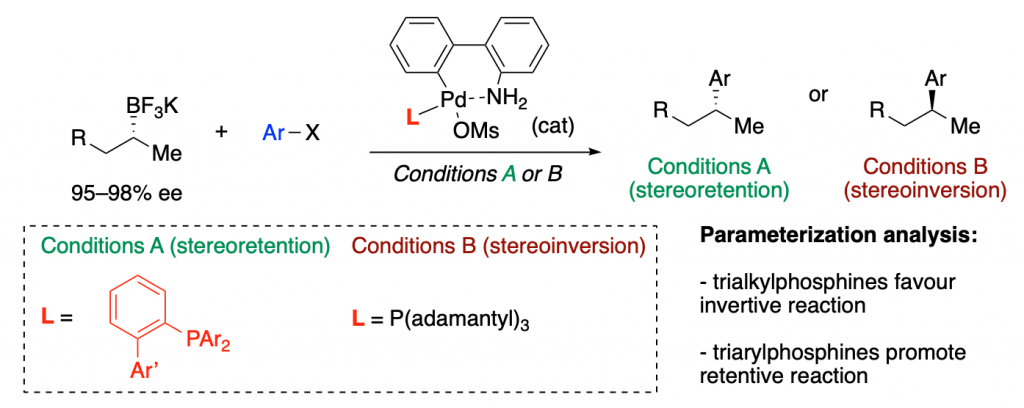
Holistic Predictions of Enantioselectivity
In 2019, Reid and Sigman reported a ground-breaking report on a model for holistic predictions of enantioselectivity in asymmetric catalysis.
As we stated in the introduction, a big part of a synthetic chemist job is to scan the literature to select some reaction conditions to test on a new substrate. This is clearly a job that a well-programmed computer should do better than a human being, especially when there are hundreds or thousands of possible conditions available.
This is a field that Sigman is pioneering, and awesome trends which allow for very significant predictions have already resulted from their efforts.
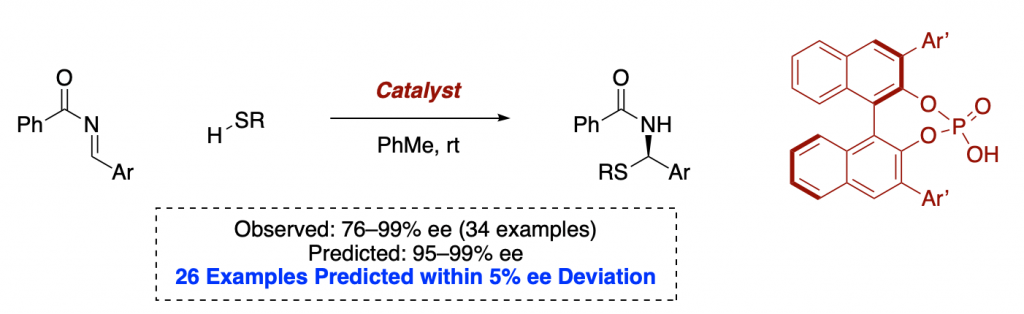
Many say that statistics, AI and machine learning could be the future of chemistry.
Machine Learning for Predicting Chemical Reactions
As Doyle group explains on their website, machine learning (which is basically statistics and computer science) can be the tool that will solve the problems of multidimensionality (which makes complex problems impossible for humans to analyze) inherent to chemical reactivity and structure.
In early 2018, Doyle reported in Science a collaborative work with Merck in which they developed a chemical model based on machine learning. They used a random forest model to predict the outcome of C–N cross coupling reactions.
Studying the Influence of Additive by Machine Learning
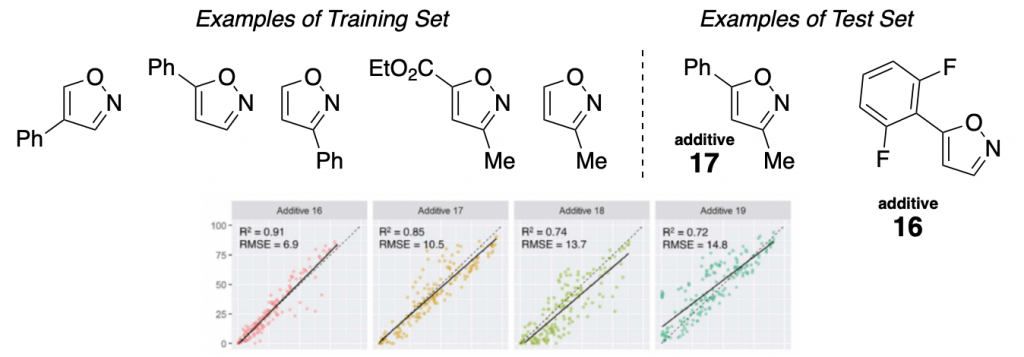
They mainly studied the influence of an additive (a family of isoxazoles) in one of the most useful reactions out there, the Buchwald-Hartwig amination.

A set of 15 different isoxazoles were used as “training set” (to obtain the linear regressions), and then another 8 of them were used as “test set”. Some examples are shown below, together with the corresponding regressions. As you can see, the data obtained with the test set correlates well with the “training regression”. Meaning that a good level of prediction is achieved.
Teaching Computers How to Do Fluorination
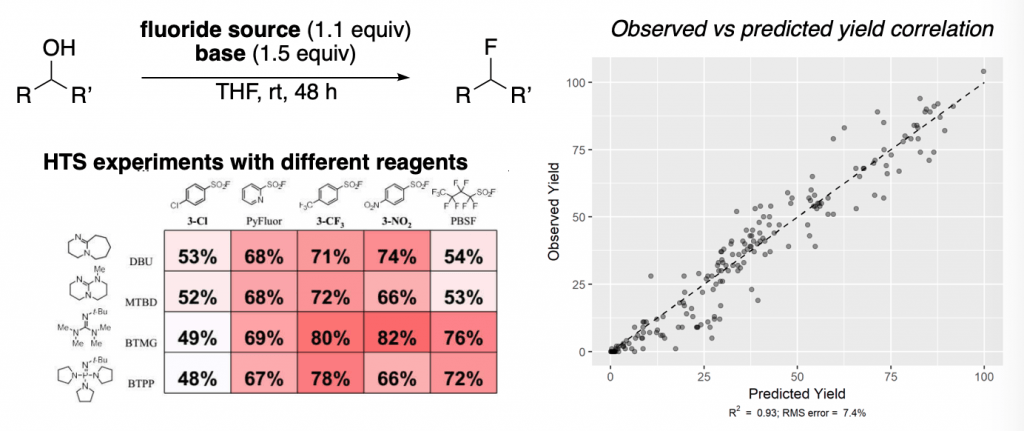
A similar concept was reported later that year by the same group, in which an awesome combination of HTS (high throughput screening) experiments and machine learning allowed developing a predictive model for the fluorination of alcohols with PyFluor. This resulted in a great expansion of the scope previously reported by Doyle and co-workers.
In the left, a schematic example of the type of HTS experiments run is displayed, showing how changes on the fluoride source and the base drastically affect the reaction yields.
The right graph shows all the results of observed yield vs. predicted yields. Very good correlations are obtained.
Are Robots Going to Take Our Jobs?
If by “our jobs” you mean by exclusively technical lab work as a chemist, the answer most likely.
But don’t get me wrong, I am nothing but optimistic about the future of chemistry. We need to embrace tools such as AI or robotics. They are here to free us from the most boring routine part of research, so we can focus in creativity to solve important problems.
On this particular matter, several research groups have been working on designing and constructing a chemical robot.
The Chemistry of the Future: Merging AI Planning with Robotic Synthesis
Timothy F. Jamison and Klavs F. Jensen from the Department of Chemistry of the Massachusetts Institute of Technology (MIT).
In 2018, they presented in Science their first version of a chemical synthesis robot. The basic idea of this machine is a complex flow chemistry system controlled by a software that allows optimizing multiple variables. So you can literally input the parameters that you want to optimize, feed the reagents, and wait until your optimization is complete. Then in a matter of days the scope of your transformation is also done.
This is how this synthesis robot looks like:

Fast-forward only one year, and this beast is where they are at:

The same MIT team published a couple of days ago a completely next-generation version of this chemical robot.
Now it is not just about chemical optimization. The synthetic system is fully integrated with an AI planning software.
This AI software is based in what we discussing over the entire article: taking data points out of thousands of published reactions, feeding them to complex algorithms, and getting optimal synthetic routes for a new or relevant target compound.
One can imagine that the third-generation of this system might even come up with its own ideas of what to synthesize. Who knows how far away we are from that…
The Future of Chemistry: Discovery Supported by Chemical Robots
The last example is mainly based upon flow chemistry systems. But some reactions are not suitable for flow. Traditional lab-scale organic synthesis is something that the Cronin group wanted to “digitalize”.
Leroy Cronin and co-workers have published in 2018 their views on how can we use algorithms to aid discovery by using chemical robots. Not long after, in early 2019, this group working at The University of Glasgow reported some of their efforts on making such robot.
Cronin’s “Chemputer” is a modular robotic platform that allows carrying out the four basic steps of organic chemistry: reaction, work-up, isolation and purification.
For this purpose, it is equipped with pumps, reactors, filtering systems, automated separatory funnels, a rotavap, and of course, software to control all the process.
The following video allows getting an idea on how this system works:
This new system based on a “chemical programming language” allowed the synthesis of several medically relevant molecules, such as sildenafil or rufinamide.
Computer-assisted Total Synthesis of Complex Natural Products
You can argue that the targets selected to test the synthetic systems described above are not of very high complexity. Typical natural product synthetic problems tackled by the big groups are much more challenging.
But computers can also assist us with those! It is just a matter of how well can we integrate DFT-based high level computations with the methods described above. This kind of integration will be relevant in the future of chemistry.
An example of such prediction is the recent synthesis of Paspaline A and Emindole PB by Timothy Newhouse and co-workers.
In this work, the authors envisioned a biosynthetic approach to those natural structures, and though of 3 possible potential intermediates. All these 3 intermediates could in principle lead to the desired natural products.
But the chosen intermediate would have to cyclize with the appropriate selectivity. In case it didn’t, the synthesis of the corresponding intermediate would have been in vain (a problem that every chemist working on natural products synthesis has encountered).
As you can see below, the proposed structures are fairly similar, it would be almost gambling for a human being to predict the outcome of each cyclization. But the structural differences make them difficult to synthesize from a common intermediate.
First Steps on the Natural Products Chemistry of the Future
To tackle this problem, Newhouse’s group predicted via DFT calculation which of the three would cyclize the way they wanted. Once they had their theoretical answer, they prepared only that intermediate precursor (saving 2/3 of the synthetic efforts required). It ended up behaving as predicted, and they completed the total synthesis.
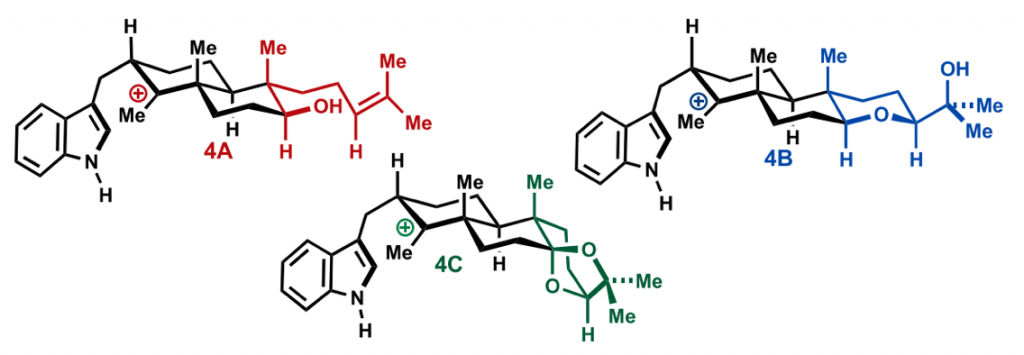
Before Newhouse, the group of Richmond Sarpong and co-workers had already applied a similar concept. They reported in 2015 the use of network-analysis to guide the retrosynthesis of very complex natural products.
Sarpong’s group applied network-analysis iteratively at the early stages of the synthetic planning of weisaconitine D and liljestrandinine, published in Nature. This allowed to come up with efficient disconnection
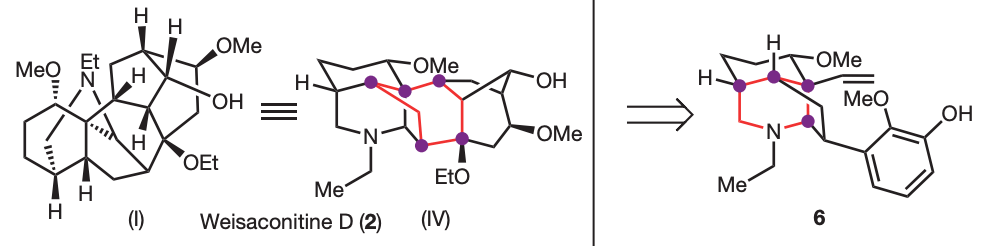
Even more recently, the same group of researchers published in J. Am. Chem. Soc. the total synthesis of the diterpenoid alkaloid arcutinidine.
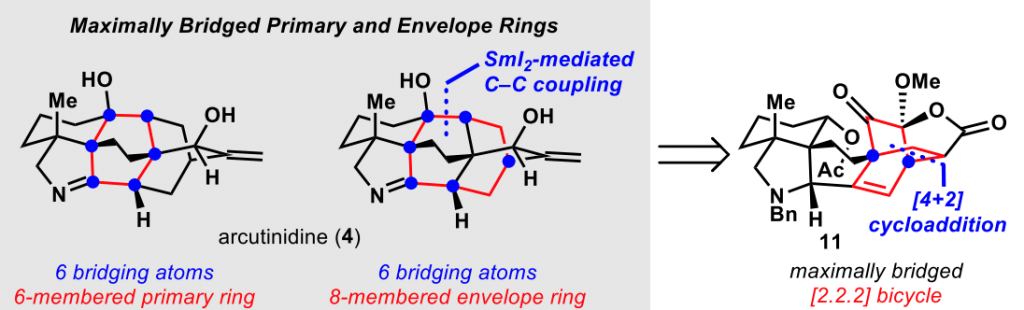
This synthesis was also aided by this network-analysis, which is inspired by the initial work already performed by E. J. Corey back in the 80s.
I wanted to close with this last set of examples, because they are a great demonstration of what, in my opinion, would be the ideal future of AI and computing in organic chemistry.
“As scientists, AI should not replace us, but rather free us from routine and boring tasks, letting us focus on what is important: solving more complex and more important problems.”
I would love to hear from your opinion on AI and computers, and how they are going to affect how we see and approach chemistry (and science in general). After all, the future of chemistry is in our hands.